ALS – Amyotrofisk lateralskleros
Amyotrofisk lateralskleros (ALS) är en grupp neurodegenerativa sjukdomar som bryter ner de nervceller som styr kroppens muskler. Sjukdomen leder till att den drabbade successivt förlorar allt fler motoriska funktioner, som förmågan att röra sig och svälja. Varje år diagnosticeras omkring 400 svenskar med sjukdomen.
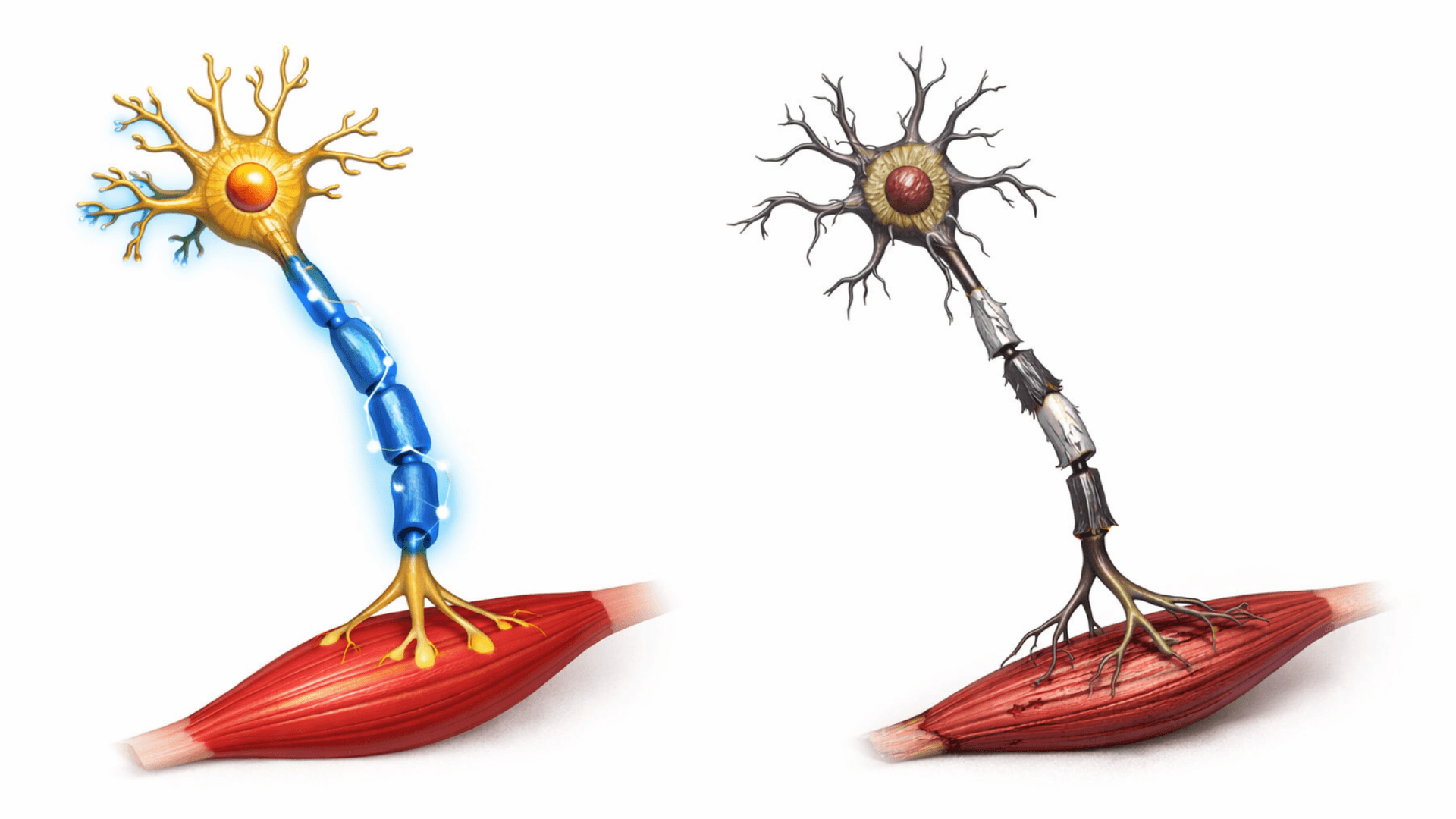
I dagsläget saknas effektiva behandlingar för ALS, med undantag för vissa former av ärftlig ALS där nya genterapier har visat lovande resultat. Sjukdomen är obotlig och de flesta patienter lever två till tre år efter att de första symtomen visar sig.
ALS i siffror
~400
svenskar får en ALS-diagnos varje år
75 %
av alla ALS-fall saknar känd orsak
10-15 %
har en ärftlig form av sjukdomen
2-3 år
typisk överlevnad efter symtomdebut
Om sjukdomen ALS
Vad är ALS?
Vid ALS angrips en typ av nervceller som kallas motorneuron. Dessa nervceller finns i hjärnbarken och i ryggmärgen, och deras uppgift är att styra våra muskler och kontrollera våra viljemässiga rörelser. När motorneuronen bryts ner förlorar hjärnan kontakten med musklerna, vilket leder till svaghet, muskelförtvining och till slut förlamning.
ALS kan drabba vem som helst, när som helst, men de flesta som insjuknar är mellan 45 och 75 år gamla. Sjukdomen är något vanligare hos män. Efter att de första symtomen uppträder är den genomsnittliga livslängden 2-3 år, men ungefär 10 procent överlever i 10 år eller längre. Förloppet kan variera kraftigt från person till person.
I dagsläget är ALS en obotlig sjukdom med dödlig utgång. Sjukvårdsinsatserna syftar främst till att förlänga livet och göra tiden som är kvar så bra som möjligt. Forskningen gör dock stora framsteg, och fler läkemedel testas nu än någonsin tidigare.
Läs mer om vad ALS är och hur sjukdomen påverkar kroppen.
Symtom och sjukdomsförlopp
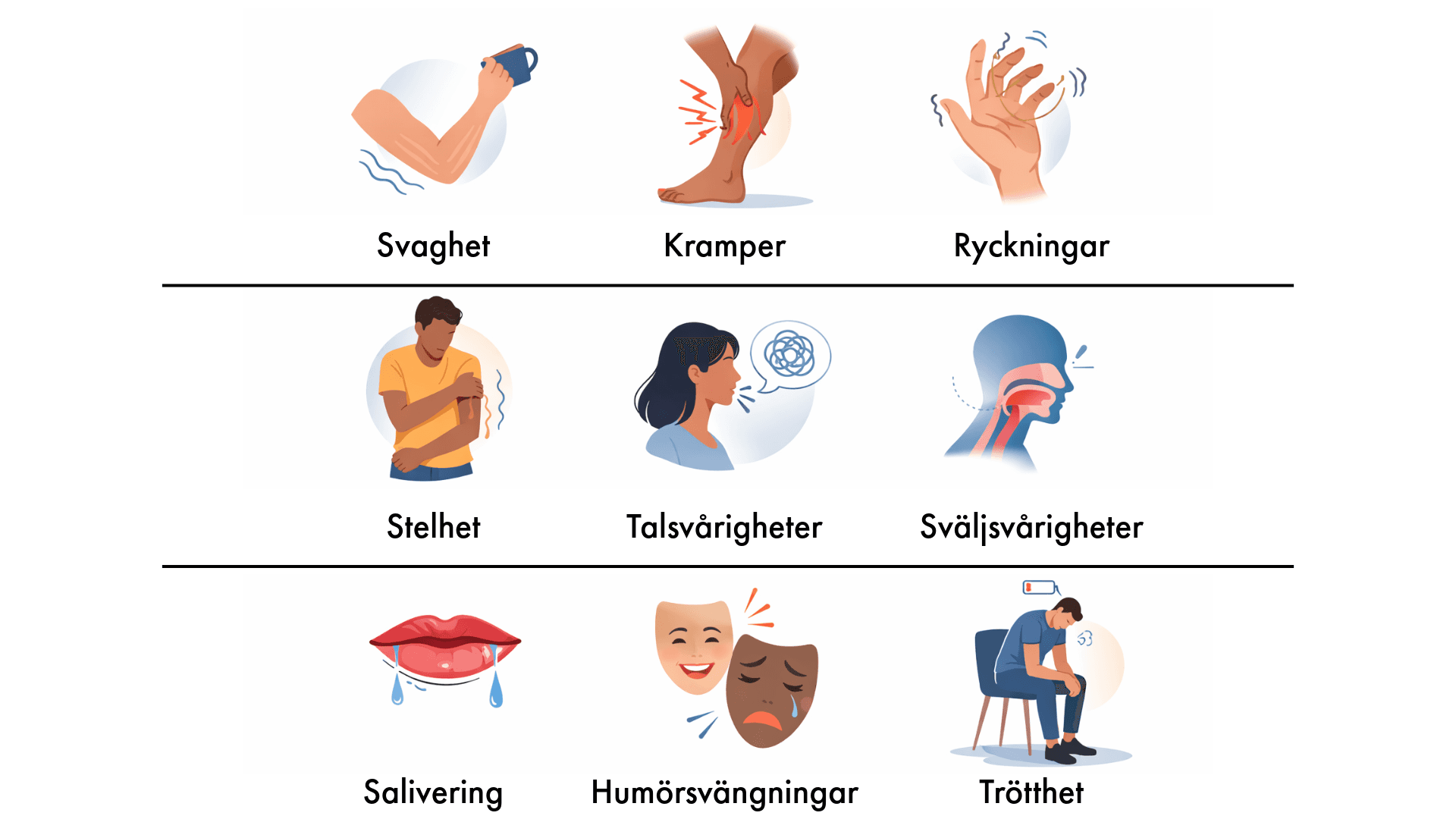
ALS börjar ofta smygande. Vanliga tidiga tecken på ALS är:
- Svaghet eller fumlighet i en hand
- Svaghet i ett ben
- Sväljsvårigheter
- Svårigheter att artikulera
- Ofrivillig viktnedgång och muskelförlust
Med tiden sprider sig svagheten i kroppen – från handen till hela armen, från armen till foten, eller tvärtom. Hastigheten varierar, men försämringen sker under veckor eller månader.
När sjukdomen börjar i en arm eller ett ben kallas det spinal ALS. När den börjar i ansikts- och svalgmuskulaturen kallas det bulbär ALS. Oavsett hur sjukdomen startar sprider den sig med tiden till hela kroppen. De flesta som avlider i ALS gör det på grund av andningssvikt, eftersom sjukdomen till slut påverkar andningsmuskulaturen.
Läs mer om första symtomen och sjukdomsförlopp.
Diagnos och behandling
ALS är en klinisk diagnos, vilket innebär att inget enskilt prov kan bekräfta eller utesluta sjukdomen. Det är den samlade bedömningen av symtom, sjukdomshistoria och undersökningsfynd som avgör. Under utredningen gör man i regel magnetkameraundersökning, lumbalpunktion, blodprover och elektromyografi. Vägen till diagnos kan ta tid, eftersom läkarna först behöver utesluta andra möjliga orsaker till symtomen.
Den enda bromsmedicinen som ervjuds svenska ALS-patienter är Riluzol (Rilutek). Medicinen introducerades på 90-talet och har endast en blygsam livsförlängande effekt.
Forskningen har dock gjort framsteg, särskilt inom genterapi. Tofersen är ett exempel på en ny behandling riktad mot en specifik genetisk mutation (SOD1) som orsakar en del av de ärftliga ALS-fallen. Fler läkemedelskandidater testas nu i kliniska studier än någonsin tidigare.
Läs mer om hur ALS diagnosticeras och om kliniska läkemedelsstudier.
Orsaker och ärftlighet
Orsaken till ALS är i de flesta fall okänd. Hos cirka 10–15 procent av alla patienter finns en tydlig ärftlighet, det vill säga en nära släkting som också har drabbats av sjukdomen. Denna form kallas ofta för familjär ALS. Övriga fall, där ingen känd ärftlighet finns, kallas då sporadisk ALS.
Tre gener har blivit särskilt förknippade med ärftlig ALS: C9orf72, SOD1 och FUS. Bärare av mutationer i dessa gener har ofta en hög risk att insjukna. Det pågår forskning på genterapier som syftar till att stänga av de muterade generna – behandlingar som i framtiden kanske kan ges till kända bärare innan de hunnit utveckla symtom.
Utöver genetiska faktorer studeras även miljöfaktorer, men inga tydliga riskfaktorer har kunnat fastslås. Forskningen pågår för att förstå vad som utlöser sjukdomen.
Läs mer om att testa sig för ärftlighet.
Påverkan på kognition och beteende
ALS påverkar inte bara rörelseförmågan. Ungefär hälften av alla patienter upplever förändringar i kognition eller beteende. Det kan yttra sig som svårigheter att hitta ord, planera aktiviteter eller kontrollera känsloyttringar. Oftast är dessa förändringar lindriga, men i vissa fall kan de vara mer uttalade. En mindre grupp patienter utvecklar utöver sin ALS även frontallobsdemens, vilket ger mer omfattande kognitiva svårigheter.
Läs mer om beteende och kognition vid ALS.
Palliativ vård
Palliativ vård syftar till att lindra symtom och ge så hög livskvalitet som möjligt under den tid som återstår. Det är naturligt att känna oro inför slutet, men det kan vara en tröst att veta att de flesta med ALS somnar in lugnt och stilla utan smärta.
Läs mer om palliativ vård vid ALS.
Leva med ALS
Att få en ALS-diagnos förändrar livet i grunden – för den som drabbas och för alla i omgivningen. Men livet tar inte slut vid en diagnos. Med rätt stöd och insatser kan man upprätthålla livskvaliteten och göra vardagen så bra som möjligt.
Ett specialiserat ALS-team – bestående av läkare, sjuksköterskor, arbetsterapeuter, kuratorer, fysioterapeuter, dietister och logopeder – spelar en avgörande roll. Teamet kan erbjuda anpassade insatser, hjälpmedel och stöd i takt med att sjukdomen fortskrider.
Personlig assistans kan ge den som lever med ALS möjlighet att leva ett mer självständigt liv. Det finns också stöd att få från kommunen, bland annat hemtjänst och olika former av avlastning.
Att vara anhörig till en person med ALS innebär stora förändringar. Det finns stöd och strategier som kan underlätta, både för partners och för barn i familjen.
- Livskvalitet och psykisk hälsa vid ALS
- Att vara närstående till någon med ALS
- Barn och föräldraskap vid ALS
- Personlig assistans och hemtjänst
- Stöd från kommunen
Personliga berättelser
Bakom varje ALS-diagnos finns en människa med en unik historia. Här samlar vi berättelser från drabbade och deras anhöriga – om livet med sjukdomen, om vardagen och om vägen framåt. Du är inte ensam.
Läs personliga berättelser om livet med ALS.
Forskning och framtidshopp
ALS-fonden finansierar forskning med målet att hitta behandlingar som kan bromsa, stoppa och i förlängningen bota sjukdomen. Forskningen inom genterapi, stamceller och biomarkörer har gjort stora framsteg de senaste åren, och fler läkemedelskandidater testas nu i kliniska studier än någonsin tidigare.
Läs mer om forskning som ALS-fonden stödjer.
Referenser och externa källor
Senaste artiklarna om ALS

Ny isländsk studie: Läkemedlet tofersen stoppar ALS
16 februari 2026

Nya genetiska fynd kan ge ledtrådar till orsaken bakom sporadisk ALS
19 december 2025

Rapport från internationell ALS-konferens i San Diego
14 december 2025
Vanliga frågor
Bidra till en värld utan ALS
Din gåva gör verklig skillnad i kampen mot ALS.